|
Spotlight Our widgets for functional genomics use Orange, a data mining
and machine learning suite. Orange can be accessed through scripting in
Python,
or by visual programming in Orange Canvas. |
| FRI > Biolab > Supplements > Microarray Data Mining with Visual Programming > D. discoideum Example > File widget | |
File widgetThe microarray data file "dicty_800_genes_from_table07.tab" is loaded into the schema with the "File" widget. Recently opened files are listed in the pull-down combo box.
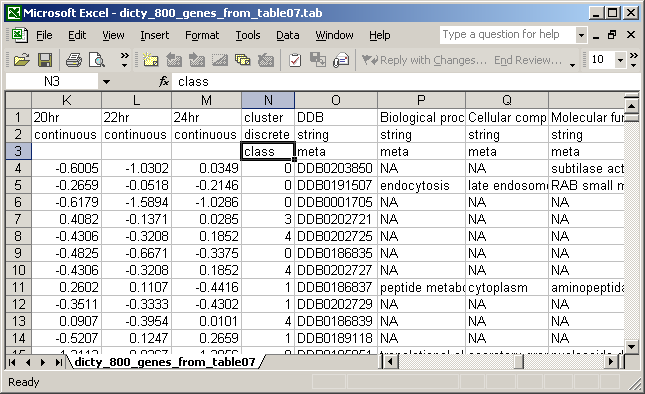
Data FormatAmong other formats, Orange can read tab-delimited files. Looking at the snapshot below one can see the required format for the tab-delimited files that can be read into schemas. The first three rows contain information about attributes (columns). The first row gives the names of attributes (20hr, 22hr,...), second row states the attribute types ("continuous" for microarray measurements, "discrete" or "string" for additional information such as annotation that can be displayed, for example in the Heat Map widget), and the third row states how the attributes should be treated. Notice that cluster ID is a special attribute called class, and will be used as such when displaying the information. Meta attributes are those that will not be considered in visualization directly, but only indirectly when displaying additional information about genes. For details on other Orange formats and for a concise definition of keywords and syntax, see Orange File Formats and a section in Orange For Beginners. The remaining rows contain the data. Each contains an expression profile of a gene, plus any additional information about the gene. The annotation is optional, since it can be added to microarray data with the GO Term Finder widget. The only required column is the one with gene names (in our case "DDB"), because this attribute is also the key in the data files with Gene Ontology annotation and genome mapping. It does not matter which name one assigns to such attribute, as widgets that handle the gene annotation or display genome map allow users to define which of the attributes holds the information on gene names. In most cases, the identification of a name attribute will be done in such widgets automatically.
|
|
 |
|